
See Our Team

-
年度 著作 2019 C.N. Chow, T.Y. Lee, Y.C. Hung, G.Z. Li, K.C. Tseng, Y.H. Liu, P.L. Kuo, H.Q. Zheng, W.C. Chang* (2019) PlantPAN3.0: a new and updated resource for reconstructing transcriptional regulatory networks from ChIP-seq experiments in plants,Nucleic Acids Research, 47(D1):D1155–D1163. 2019 W.J Zhuang, H. Chen, M. Yang, J.P Wang, Manish K. Pandey, C. Zhang, W.C. Chang, L.S. Zhang, X.T. Zhang, R.H. Tang, Vanika Garg, X.J. Wang, H.B. Tang, C.N. Chow, J.P. Wang, Ye Deng, D.P. Wang, Aamir W. Khan, Q. Yang, T.C. Cai, Prasad Bajaj, K.C. Wu, B.Z. G (2019) The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication,Nature Genetics, :. 2018 K.C. Tseng, Y.F. Chiang-Hsieh, H. Pai, C.N. Chow, S.C. Lee, H.Q. Zheng, P.L. Kuo, G.Z. Li, Y.C. Hung, N.S. Lin, and W.C. Chang* (2018) microRPM: a microRNA prediction model based only on plant small RNA sequencing data.,Bioinformatics, 34(7):1108–1115. 2018 C. N. Chow, Y. F. Chiang-Hsieh, C. H. Chien, H. Q. Zheng, T. Z. Tzong, N. Y. Wu, K. C. Tseng, P. F. Hou and W. C. Chang* (2018) Delineation of condition specific cis- and trans- acting elements in plant promoters under various endo- and exogenous stimuli,BMC Genomics, 19(Suppl 2):85. 2018 P. F. Hou, C.H. Chien, Y.F. Chiang-Hsieh, K.C. Tseng, C.N. Chow, H.J. Huang and W. C. Chang* (2018) Paddy-upland rotation for sustainable agriculture with regards to diverse soil microbial community,Scientific Reports, 8:7966. 2017 H.Q. Zheng, N.Y. Wu, C.N. Chow, K.C. Tseng, C.H. Chien, Y.C. Hung, G.Z. Li, and W.C. Chang* (2017) EXPath tool-a system for comprehensively analyzing regulatory pathways and coexpression networks from high-throughput transcriptome data.,DNA Research, 24(4):371-375. 2016 C.N. Chow, H.Q. Zheng, N.Y. Wu, C.H. Chien, H.D. Huang, T.Y. Lee, Y.F. Chiang-Hsieh, P.F. Hou, T.Y. Yang, and W. C. Chang* (2016) PlantPAN 2.0: an update of Plant Promoter Analysis Navigator for reconstructing transcriptional regulatory networks in plants.,Nucleic Acids Research, 44(D1):D1154-60. 2016 W. C. Chang, H.Q. Zheng, C. N. N. Chen (2016) Comparative transcriptome analysis reveals a potential photosynthate partitioning mechanism between lipid and starch biosynthetic pathways in green microalgae, Algal Research, 16:54-62. 2015 C.H. Chien, C.N. Chow, N.Y. Wu, Y.F. Chiang-Hsieh, P.F. Hou, and W.C. Chang* (2015) EXPath: a database of comparative expression analysis inferring metabolic pathways for plants,BMC Genomics, 16(Suppl 2):S6:page10. 2015 C.H. Chien, Y.F. Chiang-Hsieh, Y.A. Chen, C.N. Chow, N.Y. Wu, P.F. Hou, and W.C. Chang* (2015) AtmiRNET: a web-based resource for reconstructing regulatory networks of Arabidopsis microRNAs,Database (Oxford), 2015:bav042. 2014 H. Q. Zheng, Y. F. Chiang-Hsieh, C. H. Chien, J. B.K. Hsu, T. L. Liu, C. N. Chen, and W. C. Chang* (2014) AlgaePath: comprehensive analysis of metabolic pathways using transcript abundance data from next-generation sequencing in green algae,BMC Genomics, 15:196. 2014 C.L. Huang^, F.Y. Jian^, H.J. Huang^, W.C. Chang^, W.L. Wu^, C.C. Hwang*, R.H. Lee*, and T.Y. Chiang* (2014) Deciphering mycorrhizal fungi in cultivated Phalaenopsis microbiome with next-generation sequencing of multiple barcodes,Fungal Diversity, 2014:02:6. 2014 C.H. Chien, Y.F. Chiang-Hsieh, A.P. Tsou, S.L. Weng, W.C. Chang*, and H.D Huang* (2014) Large-Scale Investigation of Human TF-miRNA Relations Based on Coexpression Profiles,BioMed Research International, 2014:page8. 2013 C.T. Lu, K.Y. Huang, M.G. Su, T.Y. Lee*, N.A. Bretana, W.C. Chang, Y.J. Chen, Y.J. Chen and H.D. Huang* (2013) DbPTM 3.0: an informative resource for investigating substrate site specificity and functional association of protein post-translational modifications,Nucleic Acids Research, 41:D295-305. 2012 Y.A. Chen, Y.C. Wen, and W.C. Chang* (2012) AtPAN: an integrated system for reconstructing transcriptional regulatory networks in Arabidopsis thaliana,BMC Genomics, 13:85. 2012 T.Y. Lee*, W.C. Chang*, J. B.K. Hsu, T.H. Chang and D.M. Shien. (2012) GPMiner: an integrated system for mining combinatorial cis-regulatory elements in mammalian gene group,BMC Genomics, 13(Suppl 1):S3. 2011 C.H. Chien, Y.M. Sun, W.C. Chang, P.Y. CH, T.Y. Lee, W.C. Tsai, J.T. Horng, A.P. Tsou* and H.D. Huang* (2011) Identifying transcriptional start sites of human microRNAs based on high-throughput sequencing data,Nucleic Acids Research, 39(21):9345-9356. 2011 T.Y. Lee, J. B.K. Hsu, W.C. Chang*, and H.D. Huang* (2011) RegPhos: a system to explore the protein kinase–substrate phosphorylation network in humans,Nucleic Acids Research, 39:D777-787. 2010 Y.K. Wang, W.C. Chang, P.F. Liu, M.K. Hsiao, C.T. Lin, S.M. Lin, and R.L. Pan* (2010) Ovate family protein 1 as a plant Ku70 interacting protein involving in DNA double-strand break repair,Plant Molecular Biology, 74(4-5):453-466. 2010 T.Y. Lee, J.B.K. Hsu, F.M. Lin, W.C. Chang*, P.C. Hsu, and H.D. Huang (2010) N-Ace: using solvent accessibility and physicochemical properties to identify protein N-Acetylation sites,Journal of Computational Chemistry, 31:2759–2771. 2009 W.C. Wang, F.M. Lin, W.C. Chang, K.Y. Lin, H.D. Huang*, and N.S. Lin*. (2009) miRExpress: Analyzing high-throughput sequencing data for profiling microRNA expression,BMC Bioinformatics, 10:328. 2009 C.H. Chou^, W.C. Chang^, C.M. Chiu, C.C. Huang, H.D. Huang* (2009) FMM: a web server for metabolic pathway reconstruction and comparative analysis,Nucleic Acids Research, 37:W129-134. 2009 T.Y. Lee, J.B.K. Hsu, W.C. Chang, T.Y. Wang, P.C. Hsu, and H.D. Huang* (2009) A comprehensive resource for integrating and displaying protein post-translational modifications,BMC Research Notes, 2:111. 2009 W.C. Chang^, T.Y. Lee^, D.M. Shien, J.B.K. Hsu, J.T. Horng, P.C. Hsu, T.Y. Wang, H.D. Huang*, and R.L. Pan* (2009) Incorporating support vector machine for identifying protein tyrosine sulfation sites,Journal of Computational Chemistry, 30:2526-2537. 2009 D.M. Shien, T.Y. Lee, W.C. Chang, J.B.K. Hsu, J.T. Horng, P.C. Hsu, T.Y. Wang and H.D. Huang* (2009) Incorporating structural characteristics for identification of protein methylation sites,Journal of Computational Chemistry, 30:1532-1543. 2008 W.C. Chang, T.Y. Lee, H.D. Huang*, H.Y. Huang, and R.L. Pan* (2008) PlantPAN: Plant Promoter Analysis Navigator, for identifying combinatorial cis-regulatory elements with distance constraint in plant gene groups,BMC Genomics, 9:561. 2008 W.C. Chang, Y.K. Wang, P.F. Liu, Y.F. Tsai, L.R. Kong, C.K. Lin, C.H. Yang, and R.L. Pan* (2008) Regulation of Ku gene promoters in Arabidopsis by hormones and stress,Functional Plant Biology, 35:265-280. 2008 P.F. Liu, W.C. Chang, Y.K. Wang, H.Y. Chang, and R.L. Pan* (2008) Signaling pathways mediating the suppression of Arabidopsis thaliana Ku gene expression by abscisic acid,Biochimica et Biophysica Acta-Gene Structure and Expression, 1779:164-174. 2008 P.F. Liu, Y.K. Wang, W.C. Chang, H.Y. Chang, and R.L. Pan* (2008) Regulation of Arabidopsis thaliana Ku genes at different developmental stages under heat stress,Biochimica et Biophysica Acta-Gene Structure and Expression, 1779:402-407. 2007 P.F. Liu, W.C. Chang, Y.K. Wang, S.B. Munisamy, S.H. Hsu, H.Y. Chang, S.H. Wu, and R.L. Pan* (2007) Differential regulation of Ku gene expression in etiolated mung bean hypocotyls by auxins,Biochimica et Biophysica Acta-gene Structure and Expression, 1769:443-454. 2007 Y.Y. Hsiao, Y.J. Pan, S.H. Hsu, Y.T. Huang, T.H. Liu, C.H. Lee, P.F. Liu, W.C. Chang, Y.K. Wang, L.F. Chien, R.L. Pan* (2007) Functional roles of arginine residues in mung bean vacuolar H+-pyrophosphatase,Biochimica et Biophysica Bcta-Bioenergetics, 1767:965–973. 2004 K.H. Yan, P.F. Liu, H.T. Tzeng, W.C. Chang, W.G. Chou, R.L. Pan* (2004) Characterization of DNA end-binding activities in higher plants,Plant Physiology and Biochemistry, 8585:617–622. -

How does it work?
Transcription factors (TFs) are sequence-specific DNA-binding proteins acting as critical regulators of gene expression.
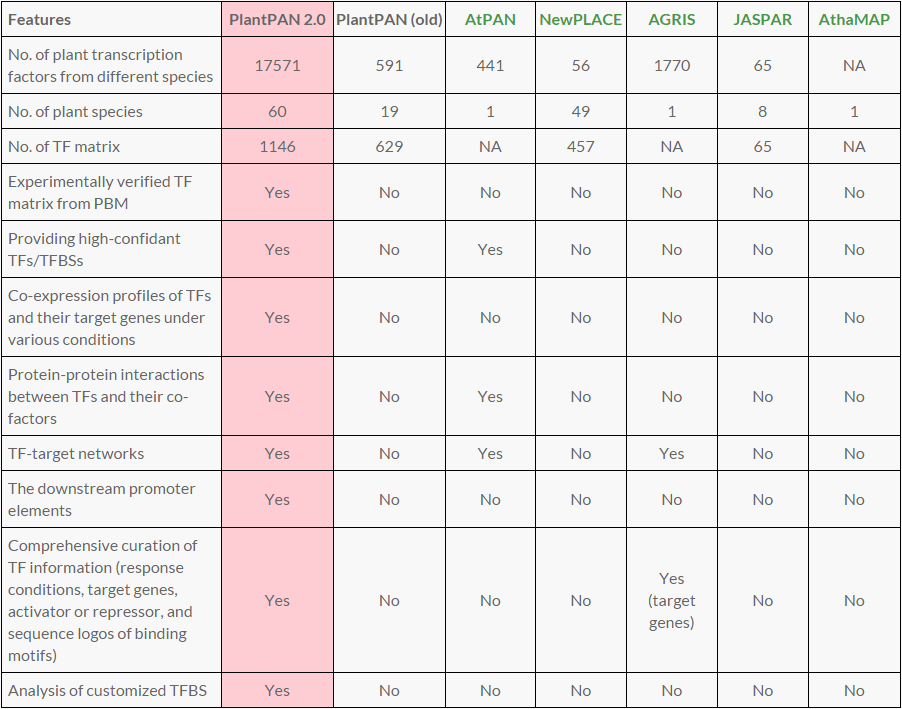
The Plant Promoter Analysis Navigator (PlantPAN; http://PlantPAN2.itps.ncku.edu.tw/) provides an informative resource for detecting transcription factor binding sites (TFBSs), corresponding TFs, and other important regulatory elements (CpG islands and tandem repeats) in a promoter or a set of promoters in plants. Additionally, TFBSs, CpG islands, and tandem repeats in the conserve regions between homologous gene promoters are also identified. The current PlantPAN release (version 2.0) contains 16,960 TFs and 1,143 matrices of TF binding sites among 76 plant species. Besides update of the annotation information, adding experimentally verified TF matrices, and improvements in visualization of transcriptional regulatory networks, several new features and functions were first incorporated.
These features include: (i) comprehensive curation of TF information (response conditions, target genes, and sequence logos of binding motifs, etc.), (ii) co-expression profiles of TFs and their target genes under various conditions (i.e., environmental stresses, hormone treatments, and developmental stages),
(iii) protein-protein interactions among TFs and their co-factors, (iv) TF-target networks, and (v) downstream promoter elements. Furthermore, a dynamic transcriptional regulatory network under various conditions is first provided in plant resources. The PlantPAN 2.0 is the most systematic platform for plant promoter analysis and reconstructing transcriptional regulatory networks.
A comparison of PlantPAN 2.0 with previous version and the similar resources
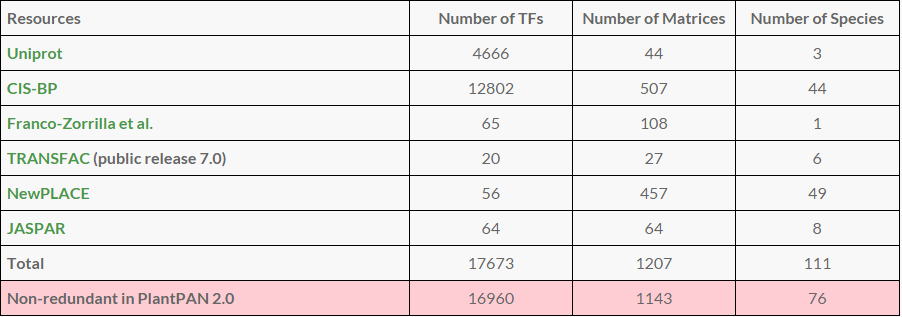
The statistics of TFs and Matrices incorporated in six resources
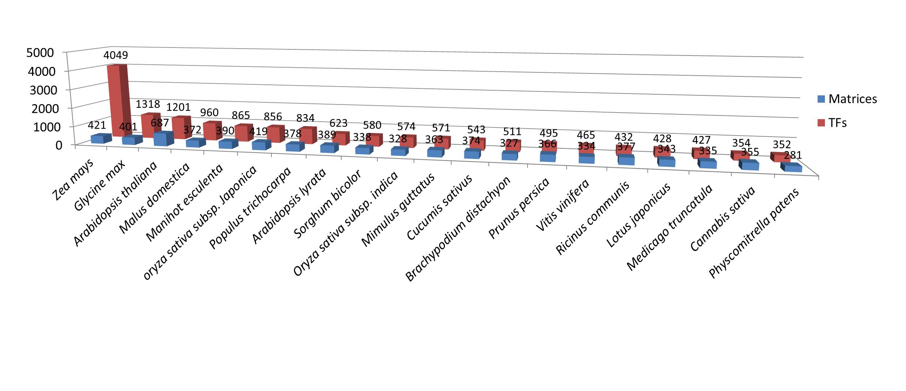
Number of TFs and matrices from top 20 species
-

Expression pathway Analysis Network
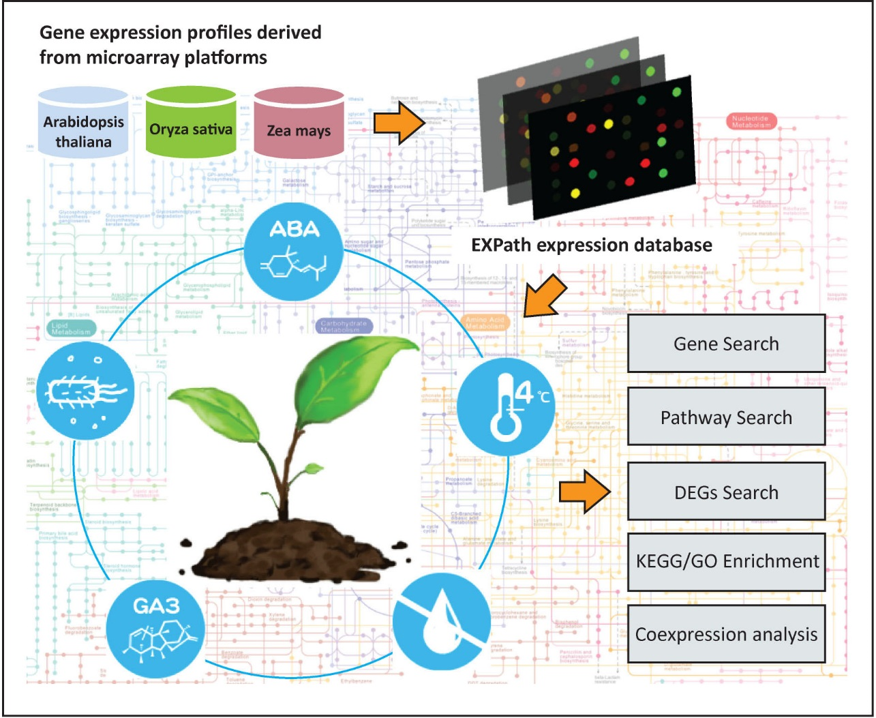
Background
In general, the expression of gene alters conditionally to catalyze a specific metabolic pathway.
Microarray-based datasets have been massively produced to monitor gene expression levels in parallel with numerous experimental treatments. Although several studies facilitated the linkage of gene expression data and metabolic pathways, none of them are amassed for plants. Moreover, advanced analysis such as pathways enrichment or how genes express under different conditions is not rendered.
Description
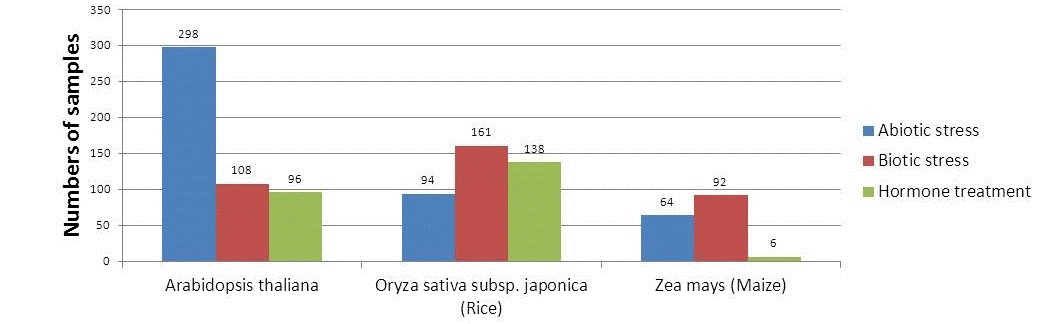
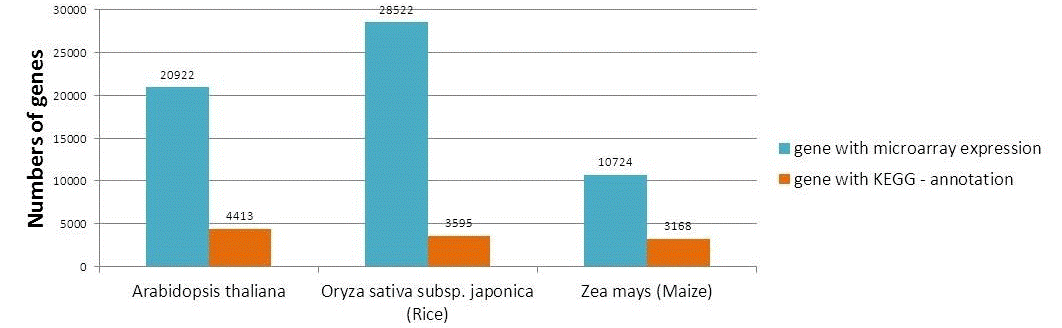
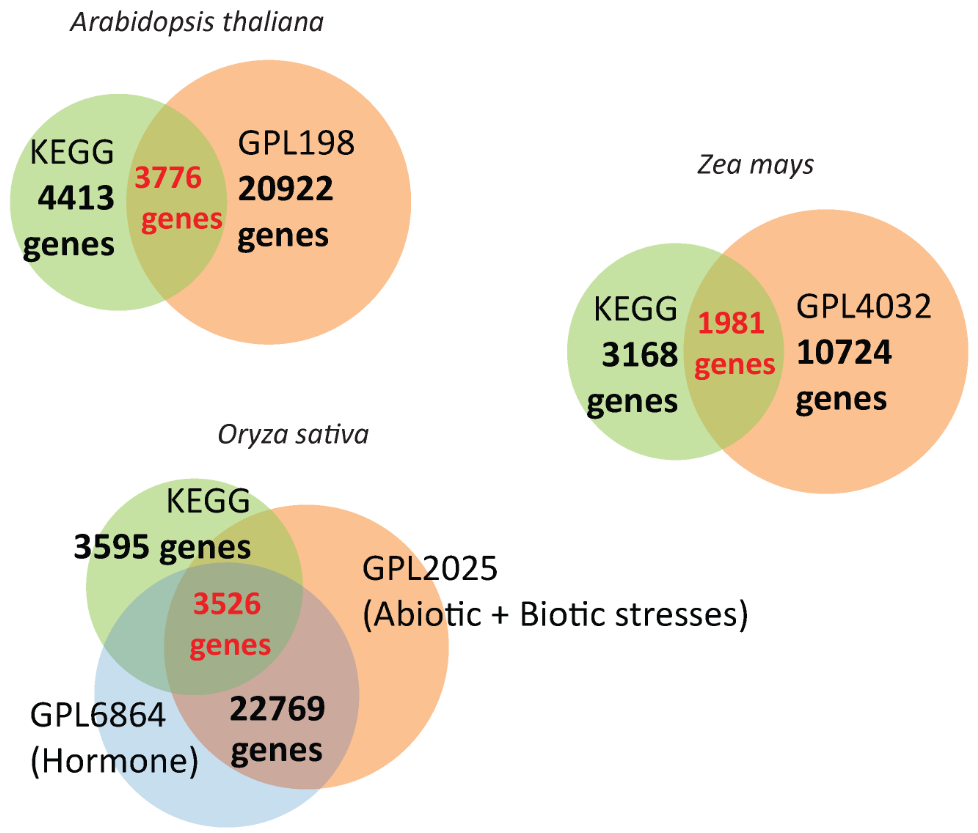
Therefore, EXPath was developed to not only comprehensively congregate the public microarray expression data from over 1000 samples in biotic stress, abiotic stress, and hormone secretion but also allow the usage of this abundant resource for coexpression analysis and differentially expression genes (DEGs) identification, finally inferring the enriched KEGG pathways and gene ontology (GO) terms of three model plants: Arabidopsis thaliana, Oryza sativa, and Zea mays. Users can access the gene expression patterns of interest under various conditions via five main functions (Gene Search, Pathway Search, DEGs Search, Pathways/GO Enrichment, and Coexpression analysis) in EXPath, which are presented by a user-friendly interface and valuable for further research.
Conclusions
In conclusion, EXPath, freely available at http://expath.itps.ncku.edu.tw, is a database resource that collects and utilizes gene expression profiles derived from microarray platforms under various conditions to infer metabolic pathways for plants.
The number of microarray samples integrated in Expath
The number of genes collected in Expath
The statistics of genes with valid expression data in EXPath
-

Arabidopsis thaliana Promoter Analysis Network
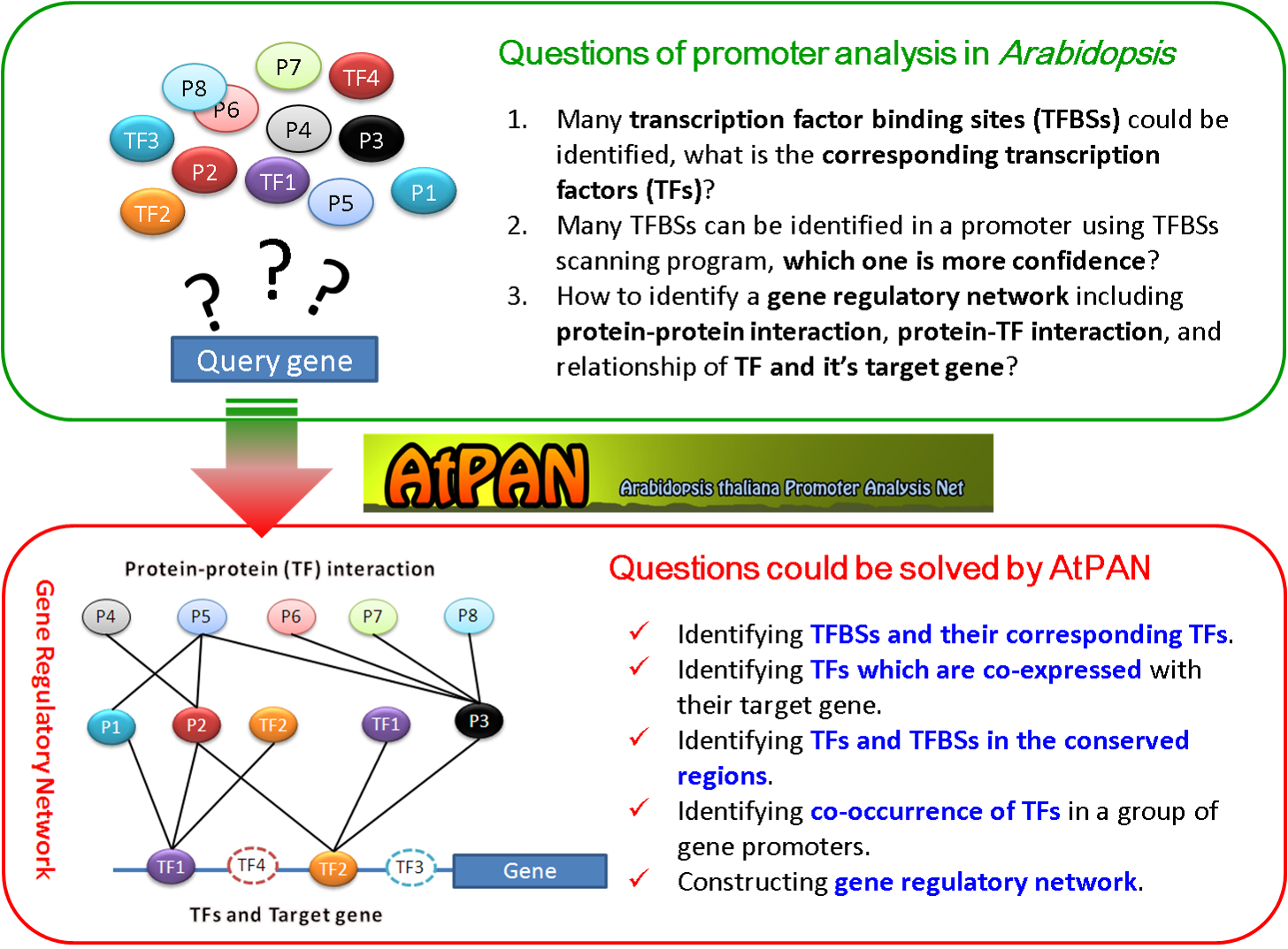
Background
Construction of transcriptional regulatory networks (TRNs) is of priority concern in systems biology. Numerous high-throughput approaches, including microarray and next-generation sequencing, are extensively adopted to examine transcriptional expression patterns on the whole-genome scale; those data are helpful in reconstructing TRNs. Identifying transcription factor binding sites (TFBSs) in a gene promoter is the initial step in elucidating the transcriptional regulation mechanism. Since transcription factors usually co-regulate a common group of genes by forming regulatory modules with similar TFBSs. Therefore, the combinatorial interactions of transcription factors must be modeled to reconstruct the gene regulatory networks.
Description
This work develops a novel database called Arabidopsis thaliana Promoter Analysis Net (AtPAN), capable of detecting TFBSs and their corresponding transcription factors (TFs) in a promoter or a set of promoters in Arabidopsis. For further analysis, according to the microarray expression data and literature, the
co-expressed TFs and their target genes can be retrieved from AtPAN. Additionally, proteins interacting with the co-expressed TFs are also incorporated to reconstruct co-expressed TRNs. Moreover, combinatorial TFs can be detected by the frequency of TFBSs co-occurrence in a group of gene promoters. In addition, TFBSs in the conserved regions between the two input sequences or homologous genes in Arabidopsis and rice are also provided in AtPAN. The output results also suggest conducting wet experiments in the future.
Conclusions
The AtPAN, which has a user-friendly input/output interface and provide graphical view of the TRNs.
-

Algae Pathway Networks
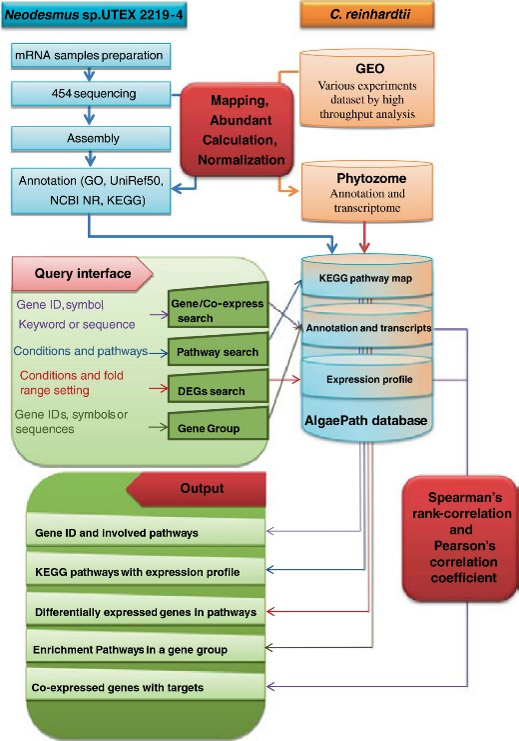
Background
Algae are important non-vascular plants that have many research applications, including high species diversity, biofuel sources, and adsorption of heavy metals and, following processing, are used as ingredients in health supplements. The increasing availability of next-generation sequencing (NGS) data for algae genomes and transcriptomes has made the development of an integrated resource for retrieving gene expression data and metabolic pathway essential for functional analysis and systems biology. In a currently available resource, gene expression profiles and biological pathways are displayed separately, making it impossible to easily search current databases to identify the cellular response mechanisms. Therefore, in this work the novel AlgaePath database was developed to retrieve transcript abundance profiles efficiently under various conditions in numerous metabolic pathways.
Description
AlgaePath is a web-based database that integrates gene information, biological pathways, and NGS datasets for the green algae Chlamydomonas reinhardtii and Neodesmus sp. UTEX 2219–4. Users can search this database to identify transcript abundance profiles and pathway information using five query pages (Gene Search, Pathway Search, Differentially Expressed Genes(DEGs) Search, Gene Group Analysis, and Co-expression Analysis). The transcript abundance data of 45 and four samples from C. reinhardtii and Neodesmus sp. UTEX 2219–4, respectively, can be obtained directly on pathway maps. Genes that are differentially expressed between two conditions can be identified using Folds Search. The Gene Group Analysis page includes a pathway enrichment analysis, and can be used to easily compare the transcript abundance profiles of functionally related genes on a map. Finally, the Co-expression Analysis page can be used to search for co-expressed transcripts of a target gene. The results of the searches will provide a valuable reference for designing further experiments and for elucidating critical mechanisms from high-throughput data.
Conclusions
AlgaePath is an effective interface that can be used to clarify the transcript response mechanisms in different metabolic pathways under various conditions. Importantly, AlgaePath can be mined to identify critical mechanisms based on high-throughput sequencing. To our knowledge, AlgaePath is the most comprehensive resource for integrating numerous databases and analysis tools in algae.
-

MicroRNA Regulatory Networks
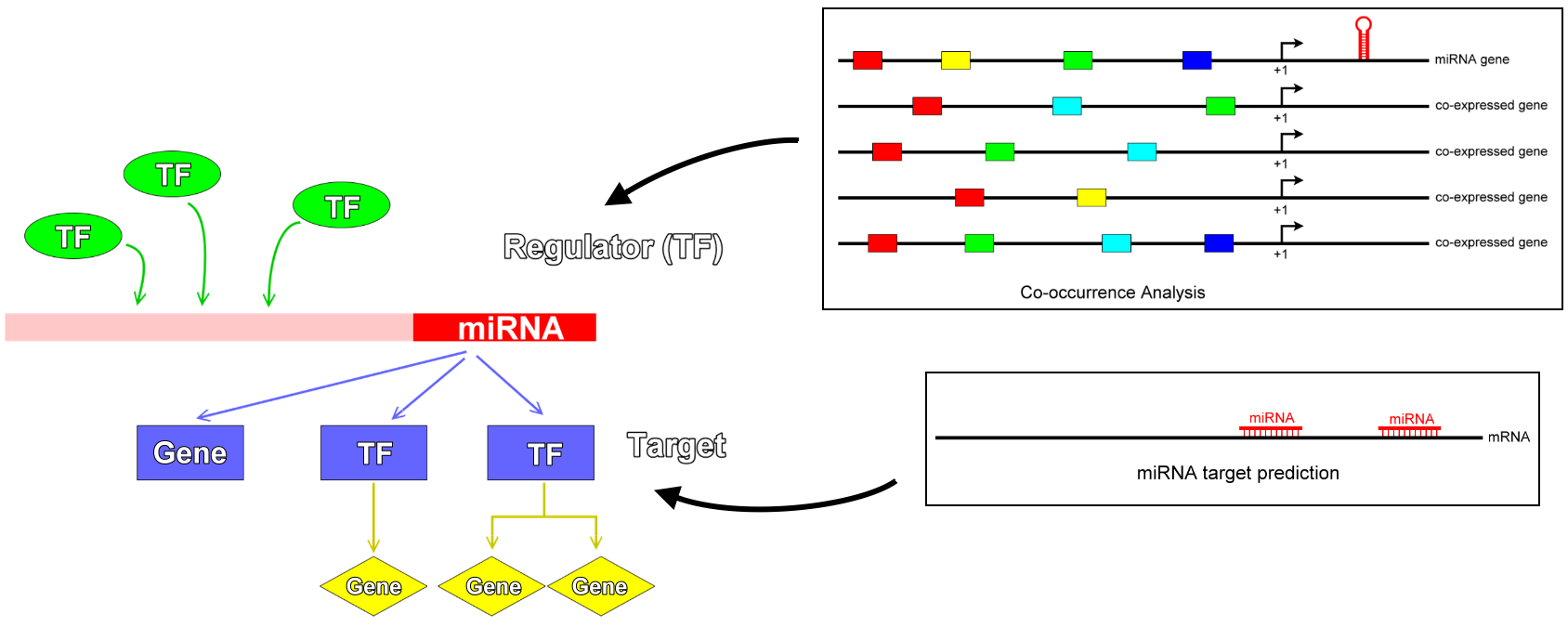
About At.miRNET
MicroRNAs are very important in post-transcriptional regulation in animals and plants.Most recent investigations in this area have focused on the targets of microRNAs; fewer have focused on their transcriptional regulation. Various databases of microRNAs target genes in mammals have been developed, but such databases in plants area limited.
No public resource integrates the upstream and downstream regulating elements of microRNAs.
These facts motivate our establishment of a database, AtmiRNet, for the retrieval of microRNA regulatory networks in Arabidopsis.Since few data about microRNA promoters are available, next-generation sequencing (NGS) data were recently used to construct a predictive model for transcription start sites (TSSs) of microRNAs in Arabidopsis based on the support vector machine (SVM) algorithm. Accordingly, in this study, 187 Arabidopsis microRNA promoter sequences were collected using 63 experimentally verified and 124 SVM-predicted TSSs.
Co-occurrence transcription factor binding sites (TFBSs) analysis was conducted to identify transcription factors (TFs) with high confidence and reconstruct upstream regulatory networks of Arabidopsis microRNAs based on co-expressed microRNAs-coding genes from microarray experimental data. Finally, several microRNAs target databases and predictive tools were integrated into AtmiRNet to develop the microRNAs regulatory networks in Arabidopsis.